|
|
|
|
Spectrometria IR, reprezinta una dintre metodele de baza ale analizei structurale, alaturi de spectroscopia de rezonanta magnetica nucleara pe care o completeaza.
Domeniul infrarosu al spectrului electromagnetic este plasat, dupa cum este aratat si in Figura IV.1, intre regiunea microundelor si regiunea vizibila, fiind cuprins in intervalul lungimilor de unda 0,8 - 200 mm. Acest domeniu corespunde din punct de vedere energetic tranzitiilor intre nivelele de vibratie ale moleculelor. Regiunea 2,5 - 15 mm reprezinta domeniul infrarosu propriu-zis si prezinta o importanta deosebita pentru analiza structurala organica. Regiunile extreme constituie infrarosul apropriat (0,8 - 2,5 mm), respectiv infrarosu indepartat (15 - 200 mm), domenii mai putin explorate in tehnica spectroscopica, dar care in anumite cazuri pot fi de interes in elucidarea structurilor moleculare.
Pozitia benzilor de absorbtie
este indicata prin lungimea de unda (![]() ) exprimata in micrometrii (mm), dar mai frecvent prin numarul de
unda (
) exprimata in micrometrii (mm), dar mai frecvent prin numarul de
unda (![]() ) in cm-1; trebuie mentionat ca se
vorbeste in mod curent despre frecventa"
unei vibratii din spectru exprimata in cm-1, fiind vorba
in realitate de numarul de unda
) in cm-1; trebuie mentionat ca se
vorbeste in mod curent despre frecventa"
unei vibratii din spectru exprimata in cm-1, fiind vorba
in realitate de numarul de unda ![]() asociat vibratiei
respective.
asociat vibratiei
respective.
Vibratia atomilor intr-o molecula poliatomica, aparent, pare o miscare dezordonata, dar se poate arata ca ea este rezultatul compunerii mai multor deplasari liniare si in faza ale nucleelor, care vor trece simultan prin pozitiile lor de echilibru si prin pozitiile extreme. Asemenea vibratii speciale constituie asa numitele moduri normale de vibratie, al caror numar este dat de numarul gradelor de libertate de vibratie. Pentru o molecula cu N atomi numarul total de grade de libertate este 3N; din acest numar daca se scad trei grade de libertate asociate miscarii de translatie de a lungul celor trei directii si trei grade de libertate pentru miscarea de rotatie in jurul celor trei axe de rotatie pentru moleculele neliniare, respectiv doua pentru cele liniare (rotatia in jurul axei interatomice avand momentul de inertie nul) se obtin 3N-6, respectiv 3N-5 grade de libertate de vibratie.
Miscarea
de vibratie este cuantificata, fiecarui mod normal de
vibratie fiindu-i asociata o energie determinata de un
numar cuantic de vibratie propriu (![]() ). Intr-o prima aproximatie energia unui nivel
molecular de vibratie va fi
data de suma celor 3N-6 (3N-5) energii.
). Intr-o prima aproximatie energia unui nivel
molecular de vibratie va fi
data de suma celor 3N-6 (3N-5) energii.
Nivelele de energie de vibratie se pot clasifica in functie de valorile pe care le au numerele cuantice de vibratie in:
nivele de zero, in care toate numerele cuantice sunt nule;
nivele fundamentale in care un singur numar cuantic este egal cu unitatea, celelalte numere cuantice fiind nule;
nivele armonice in care un singur numar cuantic este mai mare ca unitatea, restul fiind nule;
nivele de combinatie, in care mai mult de un singur numar cuantic are valoarea diferita de zero
Tranzitiile intre doua nivele de vibratie, insotite de tranzitii intre diferitele nivele de rotatie conduc la benzi de absorbtie, care constituie un spectru caracteristic fiecarui compus. Deci spectrele IR ale moleculelor poliatomice nu vor fi spectre de vibratie pure, ci spectre de vibratie-rotatie; de aceea aceste spectre nu au caracter de spectre de linii, vor fi spectre de benzi cu structura fina si de cele mai multe ori se va inregistra doar anvelopa acestei structuri.
Aceste tranzitii sunt permise numai cu
respectarea anumitor reguli de selectie. Astfel tranzitie este
permisa numai daca un singur numar cuantic de vibratie ![]() , asociat modului normal de vibratie i , variaza cu
, asociat modului normal de vibratie i , variaza cu
![]() , toate celelalte numere cuantice ramanand constante; cu
alte cuvinte la o tranzitie participa un singur mod normal de
vibratie.
, toate celelalte numere cuantice ramanand constante; cu
alte cuvinte la o tranzitie participa un singur mod normal de
vibratie.
Intensitatea benzilor de absorbtie implicand aceste tranzitii depinde de modificarea momentului de dipol datorata vibratiei. Un mod normal de vibratie va fi activ in infrarosu, numai daca acest modul normal de vibratie duce la variatia momentului de dipol al moleculei. Daca din motive de simetrie variatia momentului de dipol este nula, tranzitia care implica acel mod de vibratie este interzisa in infrarosu.
Regula de selectie ![]() , corespunde oscilatorului armonic si frecventa
cuantei absorbite ar fi egala cu frecventa proprie de vibratie.
, corespunde oscilatorului armonic si frecventa
cuantei absorbite ar fi egala cu frecventa proprie de vibratie.
Cum la temperatura camerei majoritatea moleculelor se gasesc pe nivelul de zero, datorita acestei reguli de selectie, singurele benzi observabile ar trebui sa implice numai tranzitii de pe nivelul de zero pe nivelele fundamentale, cuanta absorbita corespunzand vibratiilor fundamentale.
In spectrele IR apar insa si benzi suplimentare, care nu mai sunt datorate vibratiilor fundamentale. Astfel se pot observa armonicele frecventelor fundamentale, benzi de combinatie si benzi de rezonanta Fermi.
Armonicele frecventelor fundamentale apar la valori ale frecventelor ce sunt multiplii intregi ai celei fundamentale; dintre acestea prima armonica conduce la o banda de absorbtie relativ intensa si care poate fi usor depistata in spectru. Pe masura ce se trece la armonicele superioare intensitatea scade apreciabil. Incalcarea regulii de selectie reflecta un caracter anarmonic al vibratiei, caz in care sunt permise si variatii ale numarului cuantic diferite de unitate.
Benzile de combinatie sunt relativ slabe si apar la frecvente care reprezinta suma sau diferenta a doua sau mai multe frecvente fundamentale.
Benzile de rezonanta Fermi apar cand doua nivele vibrationale apartinand la moduri de vibratie diferite (fundamentale, armonice sau de combinatie) pot avea accidental aproximativ aceeasi energie, rezonanta dintre ele producand perturbarea acestor nivele energetice. Rezultatul va consta fie intr-o marire a intensitatii armonicei sau benzii de combinatie, fie in aparitia a doua benzi.
In functie de modificarile ce apar in molecula, cele 3N-6 moduri normale de vibratie pot sa fie: vibratii de valenta (stretching) caracterizate prin variatia lungimii de legatura, notate cu n si vibratii de deformare (bending) care duc la modificarea unghiului de valenta, notate cu d
Vibratiile de valenta si cele de deformare se pot clasifica dupa simetrie, si gradul de localizare.
Clasificarea dupa simetrie se face conform teoriei grupurilor punctuale de simetrie si permite stabilirea apartenentei fiecarui mod de vibratie la una din reprezentarile ireductibile ale grupului de simetrie de care apartine molecula; aceasta permite sa se stabileasca daca un mod de vibratie este sau nu activ in infrarosu.
In cadrul unui anumit mod de vibratie, atomii moleculei executa miscari de vibratie, cu amplitudini diferite, in jurul pozitiei de echilibru; in unele cazuri amplitudinile anumitor atomi sunt extrem de mici si acestia pot fi considerati practic imobili. In functie de numarul atomilor antrenati in vibratie, deci dupa gradul lor de localizare, vibratiile moleculelor poliatomice se pot clasifica in vibratii delocalizate si vibratii localizate
Intr-o vibratie delocalizata, atomii moleculei sunt antrenati in vibratie cu amplitudini comparabile, astfel ca intreaga molecula sau majoritatea atomilor vibreaza. Benzile de absorbtie corespunzatoare unor astfel de vibratii nu sunt caracteristice, sunt dificil de atribuit si nu sunt importante in determinarea structurii moleculare. Astfel, vibratiile unei catene de atomi de carbon, liniare sau ciclice, prezinta un caracter delocalizat si apar in toti compusii organici. Nu acesta este cazul vibratiilor localizate pe anumiti atomi ai diferitelor grupari functionale. Aceste vibratii vor fi putin influentate de restul moleculei, devenind astfel caracteristice gruparilor functionale respective si ajutand in elucidarea structurilor moleculare.
Aparitia acestor vibratii de grup se datoreaza fie diferentelor mari de masa intre atomii gruparii respective, fie unor constante de forta mult diferite intre atomii ce formeaza gruparea respectiva si intre ceilalti atomi din molecula.
Daca in cazul maselor si constantelor de forta aproximativ egale, energia de vibratie se repartizeaza uniform intre legaturi, dand nastere unei vibratii de ansamblu, delocalizate, in cazul maselor si constantelor de forta net diferite, exista un mare grad de localizare a energiei pe legatura sau legaturile gruparii respective, vibratia devenind caracteristica.
In cazul vibratiilor atomilor apartinand unei legaturi chimice (C - H, N - H, C - O , C - X, O - H, etc. ) frecventa caracteristica este data, intr-o prima aproximatie, de legea Hooke:
 (IV.13)
(IV.13)
Pozitionarea in spectru a benzii corespunzatoare unei anumite legaturi este determinata atat de constanta de forta k, cat si de masa redusa m a celor doi atomi.
Constanta de forta depinde atat de taria legaturii chimice cat si de tipul vibratiei, constanta fiind cu atat mai mare cu cat legatura este mai puternica. De asemenea vibratiile de valenta au constanta de forta mai mare decat cea de deformare:
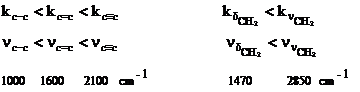 (IV.14)
(IV.14)
Pe de alta parte frecventele de vibratie vor fi cu atat mai mari cu cat masa redusa m va fi mai mica:
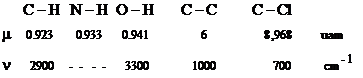 (IV.15)
(IV.15)
Vibratiile
caracteristice de valenta se pot clasifica dupa simetrie in
vibratii simetrice si asimetrice, cele asimetrice avand constanta de
forta mai mare deci si frecventa mai mare; la randul
lor vibratiile de deformare pot fi in plan sau in afara planului in cazul
gruparilor plane, sau simetrice si asimetrice in cazul
gruparilor neplanare formate din mai multi atomi (CH3, ![]() , etc.). Astfel in Figura IV.7 si IV.8 sunt
prezentate vibratiile de
valenta si de deformare ale gruparilor CH2,
respectiv CH3.
, etc.). Astfel in Figura IV.7 si IV.8 sunt
prezentate vibratiile de
valenta si de deformare ale gruparilor CH2,
respectiv CH3.
In gruparea CH2 sunt posibile
doua vibratii de valenta si patru de deformare.
Vibratiile de valenta sunt simetrice, ![]() si asimetrice,
si asimetrice, ![]() , dupa cum cei doi atomi de hidrogen oscileaza in
faza sau contrafaza. Vibratiile de deformare in plan sunt
vibratiile de forfecare
, dupa cum cei doi atomi de hidrogen oscileaza in
faza sau contrafaza. Vibratiile de deformare in plan sunt
vibratiile de forfecare ![]() (scissoring), care duc
la modificare unghiului si de leganare
(scissoring), care duc
la modificare unghiului si de leganare ![]() (rocking) in plan,
fara modificarea acestui unghi. Vibratiile in afara planului
gruparii CH2 sunt de leganare in faza deasupra
si dedesubtul planului
(rocking) in plan,
fara modificarea acestui unghi. Vibratiile in afara planului
gruparii CH2 sunt de leganare in faza deasupra
si dedesubtul planului ![]() (wagging) si de torsiune
(wagging) si de torsiune ![]() (twisting).
(twisting).
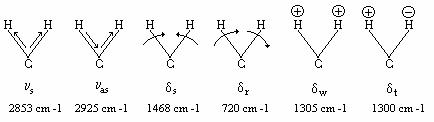
Figura IV.7 Modurile de vibratie ale gruparii metilen
In
vibratia de valenta
simetrica ![]() a gruparii metil,
cele trei legaturi C - H se alungesc si contracta in
faza, in timp ce in cea asimetrica
a gruparii metil,
cele trei legaturi C - H se alungesc si contracta in
faza, in timp ce in cea asimetrica ![]() in timp ce doua legaturi se alungesc, a treia se
contracta.
in timp ce doua legaturi se alungesc, a treia se
contracta.
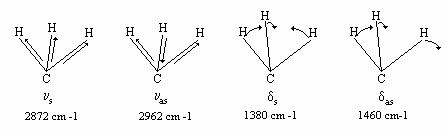
Figura IV.8 Modurile de vibratie ale gruparii metil
In
gruparea metil sunt posibile doar doua vibratii de deformare
(bending), una simetrica ![]() , in care cei trei atomi de hidrogen se aproprie simultan de
axa gruparii si cealalta asimetrica
, in care cei trei atomi de hidrogen se aproprie simultan de
axa gruparii si cealalta asimetrica ![]() , cand doi atomi de hidrogen se aproprie, iar cel de al
treilea se departeaza.
, cand doi atomi de hidrogen se aproprie, iar cel de al
treilea se departeaza.
Exista vibratii in care diferite legaturi chimice vibreaza simultan avand frecvente apropiate ca valoare; benzile de absorbtie corespunzatoare vor prezenta un caracter hibrid, ponderea fiecarei vibratii fiind determinata de amplitudinea lor relativa. Benzile hibride apar de exemplu in cazul spectrelor amidelor, alcoolilor, esterilor, etc. Astfel, in cazul amidelor, vibratia de intindere a gruparii carbonil (1597-1672 cm-1) este cuplata cu vibratia de valenta a legaturii C - N si cu cea de deformare in plan a legaturii N - H in proportie de 80%, 10% si respectiv 10%. Caracterul hibrid al benzilor nu presupune existenta efectiva a frecventelor asociate vibratiilor respective; acestea au, in general, caracter de vibratii de grup dar din punct de vedere pur calitativ ele pot fi descrise ca amestecuri de vibratii ale atomilor diferitelor legaturi.
Daca in molecula doua grupari identice sunt apropiate, ca de exemplu cele doua grupari carbonil prezente in anhidridele acizilor carboxilici, vibratiile celor doua grupari se cupleaza si are loc o despicare a benzii. Frecventele celor doua benzi corespund unor vibratii simetrice in faza si a respectiv a unor vibratii in contra faza.
Spectrometria in infrarosu isi gaseste cea mai larga intrebuintare in domeniul chimiei organice, nu numai in determinarea structurilor moleculare, dar si in elucidarea mecanismelor de reactie, in studiul cineticii de reactie si a echilibrelor chimice. Spectroscopia IR ofera in acelasi timp metode eficiente in controlul puritatii unor substante organice, precum si dozarea acestora.
In interpretarea spectrelor IR foarte importanta este pozitia, intensitatea si forma benzilor de absorbtie care pot da informatii pretioase cu privire la tipurile de legaturi si grupari functionale existente in molecula, la efectele electronice, sterice, polare, care se manifesta in cadrul structurii moleculare sau in cadrul interactiilor intermoleculare, la puritatea si concentratia unui compus intr-un amestec.
Datorita marii complexitati a spectrelor IR ale moleculelor poliatomice, atribuirea si interpretarea majoritatii benzilor din spectru nu este posibila din punct de vedere teoretic; aceasta nu scade din valoarea spectroscopiei IR in elucidarea structurii moleculare, deoarece extrem de vastul material experimental a condus la alcatuirea unor baze de date, punandu-se in acest fel bazele unei spectrometrii empirice in infrarosu. Studierea spectrelor IR au condus la concluzia ca, exceptand antipozii optici, nu exista compusi organici care sa aiba spectre IR identice; aceasta inseamna ca identitatea a doua substante chimice poate fi stabilita pe baza identitatii spectrelor lor IR.
Domeniul spectral sub 1500 cm-1 reprezinta asa numita regiune a "amprentei digitale" (fingerprint region) si este regiunea care serveste la stabilirea identitatii unui compus chimic, desi atribuirea benzilor din aceasta regiune este de cele mai multe ori dificila, daca nu imposibila.
Obtinerea informatiilor cu privire la structura compusilor organici se bazeaza pe corelarea empirica a prezentei in spectru a anumitor benzi de absorbtie, corespunzatoare unor frecvente caracteristice, cu prezenta in molecula a fie a unor grupari functionale, fie a unor tipuri de legaturi.
Prezenta simultana in spectru a mai multor frecvente caracteristice indica de cele mai multe ori, fara nici un dubiu, tipul compusului chimic. Corelarea acestor informatii cu informatiile furnizate de alte tipuri de spectre cum ar fi, in primul rand, cele de rezonanta magnetica nucleara (RMN), dar si cele de masa sau electronice (UV-VIS), conduc in final la elucidarea structurii moleculare si identificarea compusului.
Astfel existenta in spectru o unei
benzi caracteristice, ingusta, intensa, in regiunea 1700 cm-1,
indica prezenta in molecula a unei grupari carbonil.
Aceasta banda poate fi cu greu confundata si se datoreaza
vibratiei de valenta ![]() a gruparii
carbonil prezenta atat in compusii carbonilici (aldehide si
cetone), cat si in acizii carboxilici, anhidride si esteri. Faptul
ca banda apare intr-un domeniu in care nu se suprapune cu alte benzi, ii
confera acesteia o valoare analitica deosebita. Pozitia
benzii poate fi influentata nu numai de tipul functiunii
organice, ci si de geometria
moleculara, efecte sterice, efecte
a gruparii
carbonil prezenta atat in compusii carbonilici (aldehide si
cetone), cat si in acizii carboxilici, anhidride si esteri. Faptul
ca banda apare intr-un domeniu in care nu se suprapune cu alte benzi, ii
confera acesteia o valoare analitica deosebita. Pozitia
benzii poate fi influentata nu numai de tipul functiunii
organice, ci si de geometria
moleculara, efecte sterice, efecte
electronice, efecte de conjugare, prezenta anumitor substituenti in molecula, de natura solventului sau de prezenta legaturilor de hidrogen.
Indiferent de influenta acestor parametri, prezenta in spectru a unei benzi la 2720 cm-1 datorata vibratiei de valenta a legaturii C - H aldehidice, impreuna cu banda atribuita gruparii carbonil constituie o dovada spectroscopica a existentei in molecula a unei grupari aldehidice.
Acizii carboxilici vor prezenta pe
langa banda ![]() , o banda caracteristica gruparii O - H ; datorita
prezentei legaturilor de hidrogen, banda gruparii hidroxil are o forma caracteristica in
functie de concentratie: pe langa banda ingusta
datorata gruparilor O - H libere, neasociate, in zona 3500-3570 cm-1,
prezenta la dilutii mari in solventi nepolari, vibratia
caracteristica gruparilor O - H asociate prin legaturi de hidrogen se manifesta printr-o
banda larga (datorita suprapunerii benzilor corespunzatoare
diferitelor grade de asociere) in domeniul 2500-3000 cm-1.
, o banda caracteristica gruparii O - H ; datorita
prezentei legaturilor de hidrogen, banda gruparii hidroxil are o forma caracteristica in
functie de concentratie: pe langa banda ingusta
datorata gruparilor O - H libere, neasociate, in zona 3500-3570 cm-1,
prezenta la dilutii mari in solventi nepolari, vibratia
caracteristica gruparilor O - H asociate prin legaturi de hidrogen se manifesta printr-o
banda larga (datorita suprapunerii benzilor corespunzatoare
diferitelor grade de asociere) in domeniul 2500-3000 cm-1.
In cazul anhidridelor lipsa benzii caracteristice gruparilor O - H este insotita de o despicare a benzii carbonil, despicare datorata prezentei in molecula, a doua grupari carbonil, care prin cuplare pot vibra in faza sau faza contrara.
Prezenta in spectru a doua benzi relativ inguste deosebit de intense in zona 1000-1300 cm-1 atribuite legaturilor C - OsiO - R din gruparea esterica, impreuna cu banda gruparii carbonil va fi caracteristica esterilor.
Informatiile privind structura moleculara se pot obtine atat prin consultarea unor tabele de frecvente caracteristice, fie prin folosirea unor programe specializate care fac o analiza a spectrului si prin compararea cu spectrele si datele gasite in baza de date, ofera alternative cu diferite ponderi de probabilitate privind structura moleculara.
Un loc deosebit in analiza structurala organica il reprezinta, prin intermediul spectrometriei IR, studiul legaturilor de hidrogen si al echilibrelor chimice, cum ar fi tautomeria enol-cetona, lactim-lactama ,sau amino-imino.
Datorita faptului ca vibratia de valenta a gruparii hidroxil din compusii hidroxilici prezinta in spectru IR doua benzi de absorbtie (Figura IV.9), una pozitionata in zona 3600-3650 cm-1, atribuita gruparii O - H neasociate, si o alta, larga, in domeniul 3200-3400 cm-1, corespunzatoare gruparilor hidroxil implicate in legaturi de hidrogen, largimea benzii datorandu-se diferitelor grade de asociere, studiul benzilor respective aduce contributii insemnate in determinarea configuratiei si conformatiei compusilor organici.
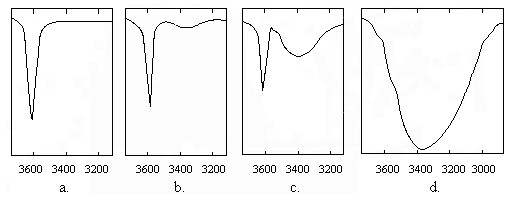 Figura IV.9 Banda atribuita gruparii hidroxil neasociate si
asociata prin
Figura IV.9 Banda atribuita gruparii hidroxil neasociate si
asociata prin
legaturi de hidrogen in ciclohexanol: a - solutie 0,03 m
in CCl4; b - solutie 0,05 m in CCl4; c - solutie 0,08 m
in CCl4; d - film lichid;
In solutii diluate, de solventi nepolari, cand formarea legaturilor de hidrogen este foarte putin probabila, in spectru apare doar banda caracteristica vibratiei de valenta O - H nelegat (Figura IV.9a), pe cand in solutii din ce in ce mai concentrate (Figura IV.9b,c), si in faza solida, sau in filme de lichid (Figura IV.9d), legaturile de hidrogen se formeaza cu usurinta si banda larga caracteristica vibratiei de valenta a gruparii hidroxil asociata devine pregnanta. Se considera ca banda larga, atribuita gruparii hidroxil asociata, nu ar fi decat anvelopa unui numar mare de benzi, corespunzatoare diferitelor moduri de asociere ale moleculelor de alcool. Maximul benzii este dependent de concentratie, de natura solventului si de temperatura. Se pot identifica, in functie de concentratie, trei tipuri de benzi atribuite hidroxilului liber, asociat in forme dimere si asociat in forme polimere.
Diferenta
![]() dintre frecventa gruparii O - H libera si
asociata este o masura a tariei legaturii de hidrogen.
In cazul legaturilor de hidrogen intramoleculare, acestea pot fi puse in
evidenta prin faptul ca nu depind de concentratie, forma si
intensitatea benzilor asociate gruparii O - H nu se modifica in urma
dilutiei.
dintre frecventa gruparii O - H libera si
asociata este o masura a tariei legaturii de hidrogen.
In cazul legaturilor de hidrogen intramoleculare, acestea pot fi puse in
evidenta prin faptul ca nu depind de concentratie, forma si
intensitatea benzilor asociate gruparii O - H nu se modifica in urma
dilutiei.
Formarea legaturilor de hidrogen duce la scaderea frecventei vibratiilor de valenta si la cresterea frecventei vibratiilor de deformare, insotita de o crestere a intensitatii si a semilargimii benzilor. Totodata pot antrena si activa unele vibratii inactive in IR si de asemenea conduce la ridicarea degenerarii anumitor nivele de vibratie cum ar fi, de exemplu, cazul vibratiei asimetrice dublu degenerate a gruparii N - H din R - NH2 in prezenta unui acceptor de protoni. Un alt efect al formarii legaturilor de hidrogen este si perturbarea unor vibratii care nu sunt direct implicate in aceste legaturi.
Combinatiile care contin gruparea carbonil si cel putin un atom de hidrogen in pozitie a, pot sa se enolizeze ca rezultat al transferului unui atom de hidrogen de la atomul din pozitie a la atomul de oxigen carbonilic. Acest proces conduce la stabilirea unui echilibru tautomer, deplasarea intr-un sens sau altul depinzand de natura compusului si de efectele electronice care pot conduce la stabilitatea uneia dintre forme.
Existenta in diferite forme tautomere reprezinta o caracteristica importanta a compusilor biomoleculari; astfel bazele purinice si pirimidinice pot manifesta doua tipuri de tautomerie: lactim - lactama si amino - imino. Aceste echilibre pot fi puse nu numai in evidenta, dar si caracterizate cu ajutorul spectometriei IR, ceea ce presupune tratarea cantitativa a absorbantei speciilor care participa la astfel de echilibre.
Absorbtia radiatiilor IR are loc cu respectarea legii Lambert - Beer:
![]() (IV.16)
(IV.16)
in care A este absorbanta sau extinctia
sistemului, T transmisia, I0 intensitatea luminii incidente, I
intensitatea luminii transmise, c concentratia solutiei, l grosimea
stratului solutiei in cm, iar ![]() coeficientul molar de
extinctie.
coeficientul molar de
extinctie.
Coeficientul molar de
extinctie sau absorbtivitatea molara este o marime
caracteristica compusului care
depinde de lungimea de unda si reprezinta absorbtia unei
radiatii monocromatice la trecerea printr-un strat de solutie de
concentratie 1 mol/l si grosime 1 cm. Absortivitatile molare au in mod uzual valori
cuprinse intre 10 - 100000 ![]() .
.
In domeniul solutiilor diluate in care se efectueaza masuratorile in spectrometria IR, UV si VIS, abaterile de la legea Lambert-Beer sunt nesemnificative.
Pentru determinarea absorbantei, in spectrometria IR se folosesc doua metode:
metoda masurarii inaltimii peakului utilizata cand absorbanta fondului ramane nemodificata la o serie de determinari. In acest caz transmisia I si I0 se masoara direct la lungimea de unda corespunzatoare maximului de absorbtie (Figura IV.10a) , caz in care absorbanta (IV.16) include si absorbtia fondului; prin determinarea absorbantei la diferite concentratii se obtine o curba de etalonare de forma:
![]() (IV.17)
(IV.17)
in care f este absorbanta fondului.
Din panta dreptei (IV.17) se poate determina coeficientul molar de absorbtie.
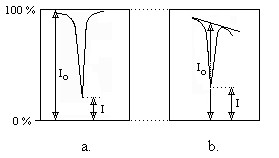
Figura IV.10Masurarea intensitatii benzilor de absorbtie in spectrul IR
metoda masurarii inaltimii peakului; b. metoda liniei de
baza;
Metoda liniei de baza utilizata cand absorbanta fondului variaza ca urmare a suprapunerii benzilor de absorbtie; in acest caz masurarea transmisiilor se face in raport cu tangenta la baza benzii de absorbtie (Figura IV.10b). Erorile care apar in aceasta metoda sunt sistematice si de aceea se folosesc si in aceasta metoda curbe de etalonare A=f(c).